
多肽药物开发新视角:非天然氨基酸与噬菌体展示技术的完美结合
2024-12-12
多肽药物作为介于生物大分子(如抗体、蛋白质)与小分子药物之间的新兴疗法,正在逐渐获得越来越多的关注。多肽既能够保持较高的特异性,又具备低成本的生产和储存优势,因此在药物开发领域中展现出独特的价值。然而,传统多肽药物开发面临一些瓶颈,例如透膜性差、血清稳定性差以及口服生物利用率低等问题。在这样的背景下,噬菌体展示(phage display)技术成为了筛选具有治疗潜力多肽的主要工具。噬菌体展示可以同时产生数十亿种多肽,但传统方法仅限于20种天然氨基酸的表达,限制了化学多样性的进一步拓展。
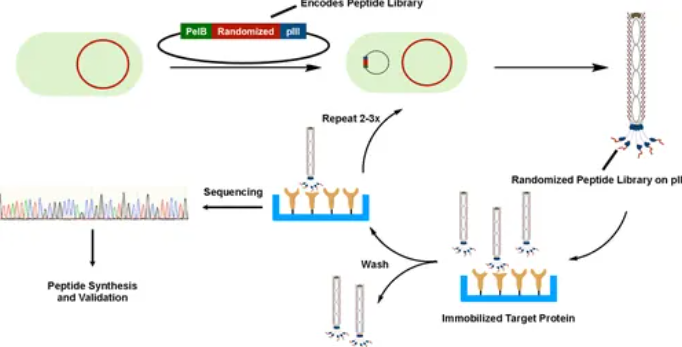
近期,美国 Texas A&M University 的 Wenshe Ray Liu 教授课题组在《Chemical Reviews》期刊中发表了一篇综述,探讨了如何通过基因编码扩展和化学修饰,将非天然氨基酸(ncAA)引入到噬菌体展示多肽库中,从而显著增加这些多肽的多样性及功能性。
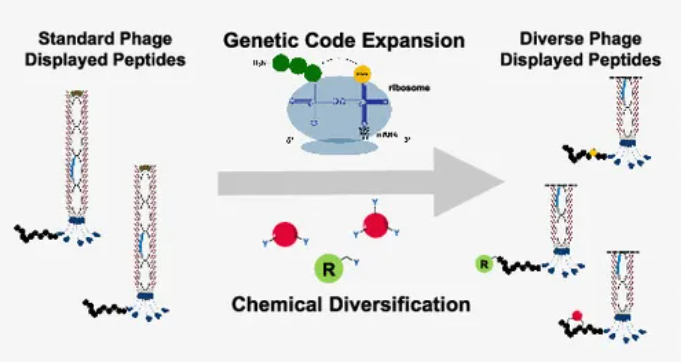
基因编码扩展在噬菌体展示中的应用
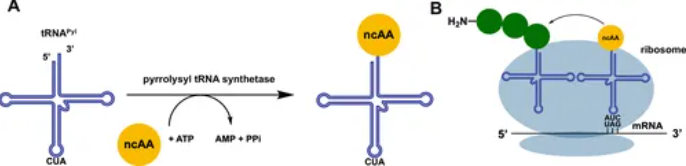
遗传编码扩展利用工程化的tRNA/tRNA合成酶对,通过琥珀(UAG)终止密码子或其他扩展编码将ncAAs整合到肽序列中。核心挑战包括确保工程化系统与天然翻译机制的正交性(不干扰天然蛋白合成)和提高ncAAs的翻译效率。
最早的研究通过琥珀终止密码子抑制成功将硒半胱氨酸(Selenocysteine)等ncAAs引入噬菌体展示系统。随后的研究开发了更广泛的tRNA合成酶对,使多种非天然氨基酸(如p-叠氮苯丙氨酸)能够成功整合到肽库中。传统琥珀抑制方法容易导致天然密码子库的寄生克隆竞争,削弱了ncAA的选择压力。近期开发的琥珀依赖肽库通过选择性富集只含有琥珀密码子的噬菌体克服了这一问题,从而提高了ncAAs整合的效率。当前,一种被广泛开发的通用密码子扩展工具是吡咯赖氨酸-tRNA合成酶/吡咯赖氨酸tRNA对(PylRS/tRNAPyl)。其中,经过突变优化的PylRS已能够识别近200种非天然氨基酸,并将其转移至tRNAPyl上,展现出广泛的应用潜力。
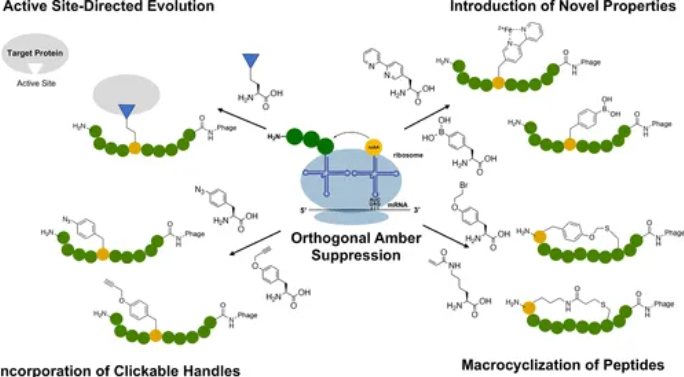
环肽因其更高的稳定性和生物活性受到关注。一些反应性ncAAs被用于在肽库中实现分子内环化,例如通过引入丙烯酰赖氨酸(AcrK)与N末端半胱氨酸反应生成环肽。成环策略是提高多肽药物特性的重要手段,主要的成环策略包括:
- 邻近反应成环:通过引入含有反应性基团的非天然氨基酸,使其能够与肽序列中的其他残基反应生成环化结构。文献中提到的丙烯酰赖氨酸(AcrK)是一种常用的非天然氨基酸,能够与肽链中的半胱氨酸发生共价反应,生成内环结构。这种环化有助于提高多肽的蛋白酶稳定性和靶标结合亲和力。
- 酰胺键成环:通过引入含有氨基和羧基的非天然氨基酸,形成肽内酰胺键环。例如,通过在肽序列中引入N端的氨基和C端的羧基,实现分子内的酰胺键形成,最终得到一个环状结构。这种策略可以有效地改善多肽的空间刚性,增加其生物活性。
- 二硫键成环:二硫键成环是自然界中常见的成环方式,通过肽链中两个半胱氨酸残基的氧化偶联,生成稳定的二硫键环化多肽。这种环化方式不仅增强了多肽的稳定性,还能增加其与特定靶标的结合能力。
- 酯键成环:除了酰胺键成环之外,酯键成环也是一种有前景的策略。通过引入含有羟基和羧基的非天然氨基酸,形成酯键环化结构。这种环化方式在提高多肽药物的透膜性和稳定性方面具有显著优势。
目前已报道的可引入非天然氨基酸总结请参考全文链接。由于正交翻译系统需要 ncAA-tRNA 复合物作为核糖体底物,许多氨基酸难以高效引入肽链,例如 α,α-双取代氨基酸、β-氨基酸、D-氨基酸、N-烷基化氨基酸(骨架修饰)及侧链带负电的氨基酸(因负电氨基酸的 tRNA 与延长因子 Tu(EF-Tu)亲和力较低)。
未来的研究方向包括改进tRNA合成酶的稳定性、扩展ncAA的种类以及实现多个ncAA的同时整合。
噬菌体展示多肽的化学修饰
化学修饰常利用肽的特定位点(如N末端和半胱氨酸残基)选择性地引入非天然基团。常用技术包括通过氧化生成醛基或酮基,然后与氨氧基化合物发生连接(如肟键连接)。例如,通过氧化生成N端醛基,再与氨氧基化合物结合,研究人员成功生成糖基化的多肽库,这些多肽能够有效识别凝集素(如DC-SIGN)。
半胱氨酸因其硫醇基的高反应性成为化学修饰的常用位点。包括:
- 马来酰亚胺修饰:半胱氨酸残基可与马来酰亚胺反应形成硫醚键,连接荧光探针或功能化分子。
- 环化反应:引入反应性非天然氨基酸(如丙烯酰赖氨酸,AcrK),并与肽中的半胱氨酸反应生成环状肽。
- 双硫键策略:在肽库中引入多个半胱氨酸,通过化学反应形成双硫键,从而生成复杂的多环肽。
在某些情况下,单一类型的化学修饰无法满足药物发现中的复杂需求。研究者们采用了多功能化策略,即在一个多肽上同时引入多种不同类型的功能基团。例如,通过遗传编码扩展引入一个ncAA,随后通过点击化学功能化,生成带有多个特定功能团的肽,以提高药物筛选效率。
未来展望
本文综述了将非天然氨基酸引入到噬菌体展示多肽库中的不同方法,并展现了这些方法在提升多肽药物筛选效率和多样性方面的巨大潜力。然而,未来仍有一些挑战需要克服,例如进一步提高遗传编码扩展的效率、优化化学修饰的选择性等。此外,如何将这些多肽药物应用于临床,解决其药物代谢和输送问题,也是未来的重要研究方向。通过进一步结合基因扩展与化学修饰手段,多肽药物的发现与开发有望迈入一个新的阶段,能够针对多种疾病靶标开发出更加高效且特异性的治疗药物。
科生景肽KPDS™平台介绍
科生景肽KPDS™是一个多功能发现平台,它集成了高度多样化的肽库建立,高通量筛选命中化合物以及人工智能/结构生物学辅助的药物化学修饰和结构优化的技术。通过采用噬菌体展示技术,从十万亿个分子中,高效筛选对靶标蛋白具有高结合力和选择性的特异性肽,再通过修饰及环化等领先的技术优势对先导多肽进行优化和改造,最终得到候选特异性靶向肽化合物,助力新药开发。这些文库可以针对几乎任何感兴趣的生物学靶标进行筛选,以高达>95%成功率,最快至1个月的速度发现命中化合物。目前已完成10余个靶标的筛选,发现几十个nM级亲和力的靶向多肽分子,覆盖肿瘤、中枢神经系统、免疫、新血管疾病等适应症。
上一页
合肥科生景肽生物科技有限公司
地址:安徽省合肥市高新区孔雀台路国家健康大数据产业园A2楼
电话:0551-65120828 0551-65120826
邮箱:inquiry@ks-vpeptide.com
联科

联科

